Chapter 4 Contradictory objectives results
Here we present the results for the satisfactory trait corverage and activation gene coverage generated by each selection scheme replicate on the contradictory objectives diagnostic. Note both of these values are gathered at the population-level. Activation gene coverage refers to the count of unique activation genes in a given population; this gives us a range of integers between 0 and 100. Satisfactory trait coverage refers to the count of unique satisfied traits in a given population; this gives us a range of integers between 0 and 100.
4.2 Satisfactory trait coverage
Satisfactory trait coverage analysis.
4.2.1 Coverage over time
Satisfactory trait coverage over time.
# data for lines and shading on plots
lines = filter(cc_over_time, diagnostic == 'contradictory_objectives') %>%
group_by(`Selection\nScheme`, gen) %>%
dplyr::summarise(
min = min(pop_uni_obj),
mean = mean(pop_uni_obj),
max = max(pop_uni_obj)
)## `summarise()` has grouped output by 'Selection Scheme'. You can override using
## the `.groups` argument.ggplot(lines, aes(x=gen, y=mean, group = `Selection\nScheme`, fill =`Selection\nScheme`, color = `Selection\nScheme`, shape = `Selection\nScheme`)) +
geom_ribbon(aes(ymin = min, ymax = max), alpha = 0.1) +
geom_line(size = 0.5) +
geom_point(data = filter(lines, gen %% 2000 == 0 & gen != 0), size = 1.5, stroke = 2.0, alpha = 1.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_continuous(
name="Generations",
limits=c(0, 50000),
breaks=c(0, 10000, 20000, 30000, 40000, 50000),
labels=c("0e+4", "1e+4", "2e+4", "3e+4", "4e+4", "5e+4")
) +
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette) +
scale_fill_manual(values = cb_palette) +
ggtitle('Satisfactory trait coverage over time')+
p_theme + theme(legend.title=element_blank(),legend.text=element_text(size=11)) +
guides(
shape=guide_legend(ncol=2, title.position = "bottom"),
color=guide_legend(ncol=2, title.position = "bottom"),
fill=guide_legend(ncol=2, title.position = "bottom")
)
4.2.2 Best coverage throughout
Best satisfactory trait coverage throughout 50,000 generations.
### best satisfactory trait coverage throughout
filter(cc_best, col == 'pop_uni_obj' & diagnostic == 'contradictory_objectives') %>%
ggplot(., aes(x = acron, y = val, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Best satisfactory trait coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=2, title.position = "bottom"),
color=guide_legend(nrow=2, title.position = "bottom"),
fill=guide_legend(nrow=2, title.position = "bottom")
)## Warning: Removed 50 rows containing missing values (`geom_point()`).
4.2.2.1 Stats
Summary statistics for the best satisfactory trait coverage.
### best
coverage = filter(cc_best, col == 'pop_uni_obj' & diagnostic == 'contradictory_objectives')
coverage$acron = factor(coverage$acron, levels = c('nds', 'lex', 'pfs', 'gfs', 'tor', 'tru', 'nov', 'ran'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(val)),
min = min(val, na.rm = TRUE),
median = median(val, na.rm = TRUE),
mean = mean(val, na.rm = TRUE),
max = max(val, na.rm = TRUE),
IQR = IQR(val, na.rm = TRUE)
)## # A tibble: 8 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 nds 50 0 85 91 91.8 100 5
## 2 lex 50 0 45 48 48.2 51 2
## 3 pfs 50 0 3 4 3.84 6 1
## 4 gfs 50 0 1 1 1 1 0
## 5 tor 50 0 1 1 1 1 0
## 6 tru 50 0 1 1 1 1 0
## 7 nov 50 0 0 0 0 0 0
## 8 ran 50 0 0 0 0 0 0Kruskal–Wallis test provides evidence of difference among satisfactory trait coverage.
##
## Kruskal-Wallis rank sum test
##
## data: val by acron
## Kruskal-Wallis chi-squared = 396.67, df = 7, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on satisfactory trait coverage.
pairwise.wilcox.test(x = coverage$val, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$val and coverage$acron
##
## nds lex pfs gfs tor tru nov
## lex <2e-16 - - - - - -
## pfs <2e-16 <2e-16 - - - - -
## gfs <2e-16 <2e-16 <2e-16 - - - -
## tor <2e-16 <2e-16 <2e-16 1 - - -
## tru <2e-16 <2e-16 <2e-16 1 1 - -
## nov <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 -
## ran <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 1
##
## P value adjustment method: bonferroni4.2.3 End of 50,000 generations
Satisfactory trait coverage in the population at the end of 50,000 generations.
### end of run
filter(cc_over_time, diagnostic == 'contradictory_objectives' & gen == 50000) %>%
ggplot(., aes(x = acron, y = pop_uni_obj, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Final satisfactory trait coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=2, title.position = "bottom"),
color=guide_legend(nrow=2, title.position = "bottom"),
fill=guide_legend(nrow=2, title.position = "bottom")
)## Warning: Removed 52 rows containing missing values (`geom_point()`).
4.2.3.1 Stats
Summary statistics for satisfactory trait coverage in the population at the end of 50,000 generations.
### end of run
coverage = filter(cc_over_time, diagnostic == 'contradictory_objectives' & gen == 50000)
coverage$acron = factor(coverage$acron, levels = c('nds', 'lex', 'pfs', 'gfs', 'tor', 'tru', 'nov', 'ran'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(pop_uni_obj)),
min = min(pop_uni_obj, na.rm = TRUE),
median = median(pop_uni_obj, na.rm = TRUE),
mean = mean(pop_uni_obj, na.rm = TRUE),
max = max(pop_uni_obj, na.rm = TRUE),
IQR = IQR(pop_uni_obj, na.rm = TRUE)
)## # A tibble: 8 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <int> <dbl> <dbl> <int> <dbl>
## 1 nds 50 0 82 86 86.4 92 3
## 2 lex 50 0 36 39 38.6 40 1
## 3 pfs 50 0 3 4 3.82 6 1
## 4 gfs 50 0 1 1 1 1 0
## 5 tor 50 0 1 1 1 1 0
## 6 tru 50 0 1 1 1 1 0
## 7 nov 50 0 0 0 0 0 0
## 8 ran 50 0 0 0 0 0 0Kruskal–Wallis test provides evidence of difference among satisfactory trait coverage in the population at the end of 50,000 generations.
##
## Kruskal-Wallis rank sum test
##
## data: pop_uni_obj by acron
## Kruskal-Wallis chi-squared = 396.7, df = 7, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on satisfactory trait coverage in the population at the end of 50,000 generations.
pairwise.wilcox.test(x = coverage$pop_uni_obj, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$pop_uni_obj and coverage$acron
##
## nds lex pfs gfs tor tru nov
## lex <2e-16 - - - - - -
## pfs <2e-16 <2e-16 - - - - -
## gfs <2e-16 <2e-16 <2e-16 - - - -
## tor <2e-16 <2e-16 <2e-16 1 - - -
## tru <2e-16 <2e-16 <2e-16 1 1 - -
## nov <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 -
## ran <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 <2e-16 1
##
## P value adjustment method: bonferroni4.3 Activation gene coverage
Activation gene coverage analysis.
4.3.1 Over time coverage
Activation gene coverage over time.
# data for lines and shading on plots
lines = filter(cc_over_time, diagnostic == 'contradictory_objectives') %>%
group_by(`Selection\nScheme`, gen) %>%
dplyr::summarise(
min = min(uni_str_pos),
mean = mean(uni_str_pos),
max = max(uni_str_pos)
)## `summarise()` has grouped output by 'Selection Scheme'. You can override using
## the `.groups` argument.ggplot(lines, aes(x=gen, y=mean, group = `Selection\nScheme`, fill =`Selection\nScheme`, color = `Selection\nScheme`, shape = `Selection\nScheme`)) +
geom_ribbon(aes(ymin = min, ymax = max), alpha = 0.1) +
geom_line(size = 0.5) +
geom_point(data = filter(lines, gen %% 2000 == 0 & gen != 0), size = 1.5, stroke = 2.0, alpha = 1.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_continuous(
name="Generations",
limits=c(0, 50000),
breaks=c(0, 10000, 20000, 30000, 40000, 50000),
labels=c("0e+4", "1e+4", "2e+4", "3e+4", "4e+4", "5e+4")
) +
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette) +
scale_fill_manual(values = cb_palette) +
ggtitle('Activation gene coverage over time')+
p_theme + theme(legend.title=element_blank(),legend.text=element_text(size=11)) +
guides(
shape=guide_legend(ncol=2, title.position = "bottom"),
color=guide_legend(ncol=2, title.position = "bottom"),
fill=guide_legend(ncol=2, title.position = "bottom")
)
4.3.2 End of 50,000 generations
Activation gene coverage in the population at the end of 50,000 generations.
# end of run
filter(cc_over_time, diagnostic == 'contradictory_objectives' & gen == 50000) %>%
ggplot(., aes(x = acron, y = uni_str_pos, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Final activation gene coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=2, title.position = "bottom"),
color=guide_legend(nrow=2, title.position = "bottom"),
fill=guide_legend(nrow=2, title.position = "bottom")
)## Warning: Removed 17 rows containing missing values (`geom_point()`).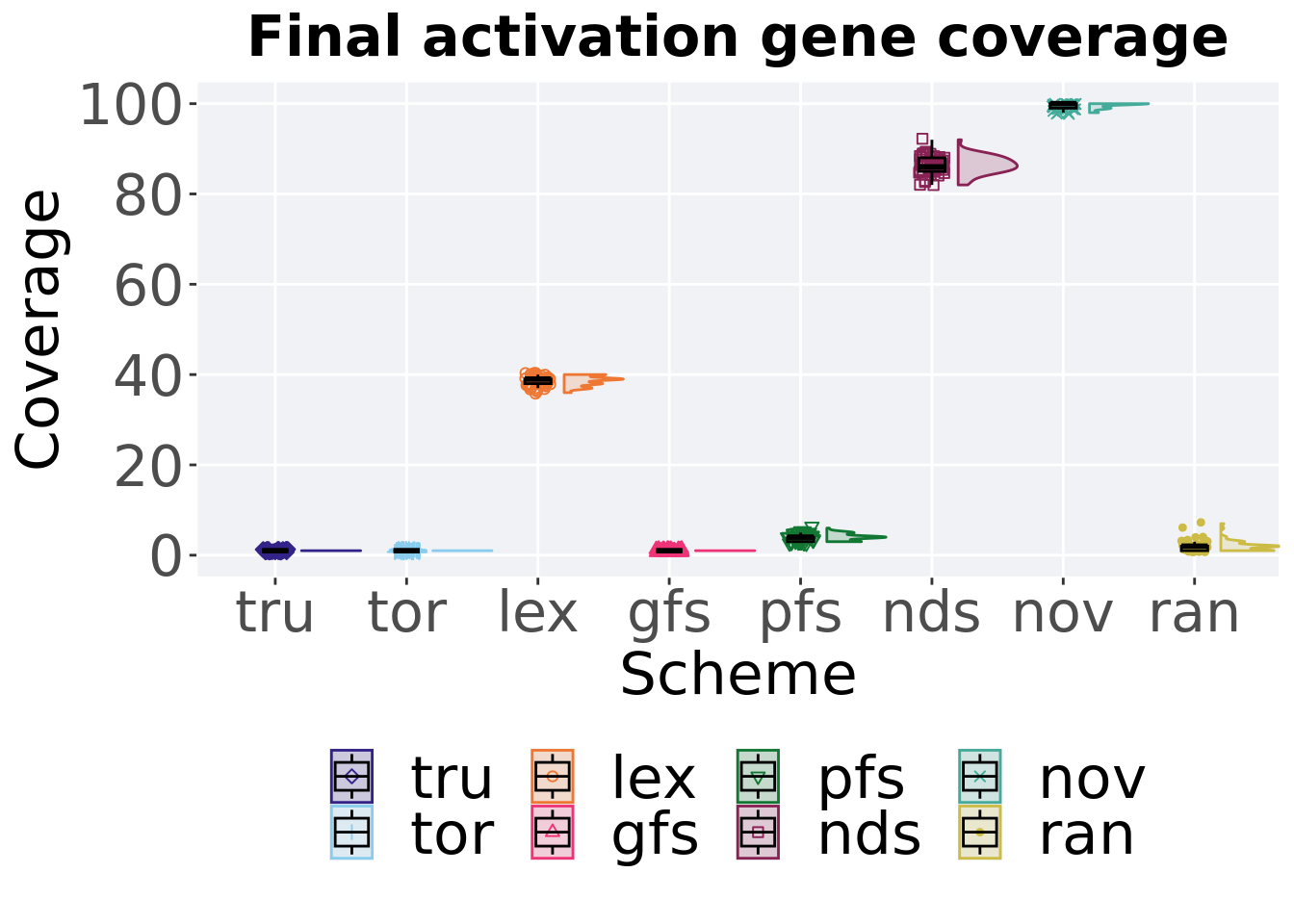
4.3.2.1 Stats
Summary statistics for activation gene coverage.
# end of run
coverage = filter(cc_over_time, diagnostic == 'contradictory_objectives' & gen == 50000)
coverage$acron = factor(coverage$acron, levels = c('nov', 'nds', 'lex', 'pfs', 'ran', 'gfs', 'tor', 'tru'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(uni_str_pos)),
min = min(uni_str_pos, na.rm = TRUE),
median = median(uni_str_pos, na.rm = TRUE),
mean = mean(uni_str_pos, na.rm = TRUE),
max = max(uni_str_pos, na.rm = TRUE),
IQR = IQR(uni_str_pos, na.rm = TRUE)
)## # A tibble: 8 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <int> <dbl> <dbl> <int> <dbl>
## 1 nov 50 0 98 100 99.6 100 1
## 2 nds 50 0 82 86 86.4 92 3
## 3 lex 50 0 36 39 38.6 40 1
## 4 pfs 50 0 3 4 3.98 6 1
## 5 ran 50 0 1 2 2.06 7 1
## 6 gfs 50 0 1 1 1 1 0
## 7 tor 50 0 1 1 1 1 0
## 8 tru 50 0 1 1 1 1 0Kruskal–Wallis test provides evidence of difference among activation gene coverage.
##
## Kruskal-Wallis rank sum test
##
## data: uni_str_pos by acron
## Kruskal-Wallis chi-squared = 384.23, df = 7, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on activation gene coverage.
pairwise.wilcox.test(x = coverage$uni_str_pos, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$uni_str_pos and coverage$acron
##
## nov nds lex pfs ran gfs tor
## nds < 2e-16 - - - - - -
## lex < 2e-16 < 2e-16 - - - - -
## pfs < 2e-16 < 2e-16 < 2e-16 - - - -
## ran < 2e-16 < 2e-16 < 2e-16 2.9e-12 - - -
## gfs < 2e-16 < 2e-16 < 2e-16 < 2e-16 3.0e-10 - -
## tor < 2e-16 < 2e-16 < 2e-16 < 2e-16 3.0e-10 1 -
## tru < 2e-16 < 2e-16 < 2e-16 < 2e-16 3.0e-10 1 1
##
## P value adjustment method: bonferroni4.4 Nondominated sorting split
Here analyze the satisfactory trait coverage and activation gene coverage results for nondominated sorting, nondominated front ranking (no fitness sharing between fronts), and phenotypic fitness sharing.
4.4.1 Coverage over time
Satisfactory trait coverage over time.
lines = filter(nss, diagnostic == 'contradictory_objectives') %>%
group_by(`Selection\nScheme`, gen) %>%
dplyr::summarise(
min = min(pop_uni_obj),
mean = mean(pop_uni_obj),
max = max(pop_uni_obj)
)## `summarise()` has grouped output by 'Selection Scheme'. You can override using
## the `.groups` argument.ggplot(lines, aes(x=gen, y=mean, group = `Selection\nScheme`, fill =`Selection\nScheme`, color = `Selection\nScheme`, shape = `Selection\nScheme`)) +
geom_ribbon(aes(ymin = min, ymax = max), alpha = 0.1) +
geom_line(size = 0.5) +
geom_point(data = filter(lines, gen %% 2000 == 0 & gen != 0), size = 1.5, stroke = 2.0, alpha = 1.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_continuous(
name="Generations",
limits=c(0, 50000),
breaks=c(0, 10000, 20000, 30000, 40000, 50000),
labels=c("0e+4", "1e+4", "2e+4", "3e+4", "4e+4", "5e+4")
) +
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette) +
scale_fill_manual(values = cb_palette) +
ggtitle('Satisfactory trait coverage over time')+
p_theme + theme(legend.title=element_blank(),legend.text=element_text(size=11)) +
guides(
shape=guide_legend(ncol=1, title.position = "bottom"),
color=guide_legend(ncol=1, title.position = "bottom"),
fill=guide_legend(ncol=1, title.position = "bottom")
)
4.4.2 Best coverage throughout
Best satisfactory trait coverage.
### best satisfactory trait coverage throughout
coverage %>%
ggplot(., aes(x = acron, y = val, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Best satisfactory trait coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=1, title.position = "bottom"),
color=guide_legend(nrow=1, title.position = "bottom"),
fill=guide_legend(nrow=1, title.position = "bottom")
)## Warning: Removed 2 rows containing missing values (`geom_point()`).
4.4.2.1 Stats
Summary statistics for the best satisfactory trait coverage.
# summary
coverage$acron = factor(coverage$acron, levels = c('nds', 'pfs', 'nfr'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(val)),
min = min(val, na.rm = TRUE),
median = median(val, na.rm = TRUE),
mean = mean(val, na.rm = TRUE),
max = max(val, na.rm = TRUE),
IQR = IQR(val, na.rm = TRUE)
)## # A tibble: 3 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 nds 50 0 85 91 91.8 100 5
## 2 pfs 50 0 1 1 1 1 0
## 3 nfr 50 0 28 44 42.8 54 8.5Kruskal–Wallis test provides evidence of difference among best satisfactory trait coverage.
##
## Kruskal-Wallis rank sum test
##
## data: val by acron
## Kruskal-Wallis chi-squared = 137.61, df = 2, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on best satisfactory trait coverage.
pairwise.wilcox.test(x = coverage$val, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$val and coverage$acron
##
## nds pfs
## pfs <2e-16 -
## nfr <2e-16 1
##
## P value adjustment method: bonferroni4.4.3 End of 50,000 generations
Satisfactory trait coverage in the population at the end of 50,000 generations.
coverage %>%
ggplot(., aes(x = acron, y = pop_uni_obj, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Final satisfactory trait coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=1, title.position = "bottom"),
color=guide_legend(nrow=1, title.position = "bottom"),
fill=guide_legend(nrow=1, title.position = "bottom")
)
4.4.3.1 Stats
Summary statistics for satisfactory trait coverage in the population at the end of 50,000 generations.
coverage$acron = factor(coverage$acron, levels = c('nds', 'pfs', 'nfr'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(pop_uni_obj)),
min = min(pop_uni_obj, na.rm = TRUE),
median = median(pop_uni_obj, na.rm = TRUE),
mean = mean(pop_uni_obj, na.rm = TRUE),
max = max(pop_uni_obj, na.rm = TRUE),
IQR = IQR(pop_uni_obj, na.rm = TRUE)
)## # A tibble: 3 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <int> <dbl> <dbl> <int> <dbl>
## 1 nds 50 0 82 86 86.4 92 3
## 2 pfs 50 0 3 4 3.82 6 1
## 3 nfr 50 0 1 1 1.28 2 1Kruskal–Wallis test provides evidence of difference among satisfactory trait coverage in the population at the end of 50,000 generations.
##
## Kruskal-Wallis rank sum test
##
## data: pop_uni_obj by acron
## Kruskal-Wallis chi-squared = 135.36, df = 2, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on satisfactory trait coverage in the population at the end of 50,000 generations.
pairwise.wilcox.test(x = coverage$pop_uni_obj, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$pop_uni_obj and coverage$acron
##
## nds pfs
## pfs <2e-16 -
## nfr <2e-16 <2e-16
##
## P value adjustment method: bonferroni4.5 Multi-valley crossing results
4.5.1 Satisfactory trait coverage
Satisfactory trait coverage analysis.
4.5.1.1 Coverage over time
Satisfactory trait coverage over time.
# data for lines and shading on plots
lines = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives') %>%
group_by(`Selection\nScheme`, gen) %>%
dplyr::summarise(
min = min(pop_uni_obj),
mean = mean(pop_uni_obj),
max = max(pop_uni_obj)
)## `summarise()` has grouped output by 'Selection Scheme'. You can override using
## the `.groups` argument.ggplot(lines, aes(x=gen, y=mean, group = `Selection\nScheme`, fill =`Selection\nScheme`, color = `Selection\nScheme`, shape = `Selection\nScheme`)) +
geom_ribbon(aes(ymin = min, ymax = max), alpha = 0.1) +
geom_line(size = 0.5) +
geom_point(data = filter(lines, gen %% 2000 == 0 & gen != 0), size = 1.5, stroke = 2.0, alpha = 1.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 5),
breaks=seq(0,5, 1)
) +
scale_x_continuous(
name="Generations",
limits=c(0, 50000),
breaks=c(0, 10000, 20000, 30000, 40000, 50000),
labels=c("0e+4", "1e+4", "2e+4", "3e+4", "4e+4", "5e+4")
) +
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette) +
scale_fill_manual(values = cb_palette) +
ggtitle('Satisfactory trait coverage over time')+
p_theme + theme(legend.title=element_blank(),legend.text=element_text(size=11)) +
guides(
shape=guide_legend(ncol=2, title.position = "bottom"),
color=guide_legend(ncol=2, title.position = "bottom"),
fill=guide_legend(ncol=2, title.position = "bottom")
)
4.5.1.2 Best coverage throughout
Best satisfactory trait coverage throughout 50,000 generations.
### best satisfactory trait coverage throughout
filter(cc_best_mvc, col == 'pop_uni_obj' & diagnostic == 'contradictory_objectives') %>%
ggplot(., aes(x = acron, y = val, color = acron, fill = acron, shape = acron)) +
geom_flat_violin(position = position_nudge(x = .2, y = 0), scale = 'width', alpha = 0.2) +
geom_point(position = position_jitter(width = .1), size = 1.5, alpha = 1.0) +
geom_boxplot(color = 'black', width = .2, outlier.shape = NA, alpha = 0.0) +
guides(fill = "none",color = 'none', shape = 'none') +
scale_y_continuous(
name="Coverage",
limits=c(0, 5)
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette, ) +
scale_fill_manual(values = cb_palette) +
ggtitle('Best satisfactory trait coverage')+
p_theme + theme(legend.title=element_blank()) +
guides(
shape=guide_legend(nrow=2, title.position = "bottom"),
color=guide_legend(nrow=2, title.position = "bottom"),
fill=guide_legend(nrow=2, title.position = "bottom")
)## Warning: Removed 178 rows containing missing values (`geom_point()`).
4.5.1.2.1 Stats
Summary statistics for the best satisfactory trait coverage.
### best
coverage = filter(cc_best_mvc, col == 'pop_uni_obj' & diagnostic == 'contradictory_objectives')
coverage$acron = factor(coverage$acron, levels = c('pfs','nds', 'lex', 'gfs', 'tor', 'tru', 'nov', 'ran'))
coverage %>%
group_by(acron) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(val)),
min = min(val, na.rm = TRUE),
median = median(val, na.rm = TRUE),
mean = mean(val, na.rm = TRUE),
max = max(val, na.rm = TRUE),
IQR = IQR(val, na.rm = TRUE)
)## # A tibble: 8 x 8
## acron count na_cnt min median mean max IQR
## <fct> <int> <int> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 pfs 50 0 2 3 3.42 5 1
## 2 nds 50 0 0 0 0 0 0
## 3 lex 50 0 0 0 0 0 0
## 4 gfs 50 0 0 0 0 0 0
## 5 tor 50 0 0 0 0 0 0
## 6 tru 50 0 0 0 0 0 0
## 7 nov 50 0 0 0 0 0 0
## 8 ran 50 0 0 0 0 0 0Kruskal–Wallis test provides evidence of difference among satisfactory trait coverage.
##
## Kruskal-Wallis rank sum test
##
## data: val by acron
## Kruskal-Wallis chi-squared = 396.91, df = 7, p-value < 2.2e-16Results for post-hoc Wilcoxon rank-sum test with a Bonferroni correction on satisfactory trait coverage.
pairwise.wilcox.test(x = coverage$val, g = coverage$acron, p.adjust.method = "bonferroni",
paired = FALSE, conf.int = FALSE, alternative = 'l')##
## Pairwise comparisons using Wilcoxon rank sum test with continuity correction
##
## data: coverage$val and coverage$acron
##
## pfs nds lex gfs tor tru nov
## nds <2e-16 - - - - - -
## lex <2e-16 1 - - - - -
## gfs <2e-16 1 1 - - - -
## tor <2e-16 1 1 1 - - -
## tru <2e-16 1 1 1 1 - -
## nov <2e-16 1 1 1 1 1 -
## ran <2e-16 1 1 1 1 1 1
##
## P value adjustment method: bonferroni4.5.1.3 Coverage comparison
Best performances in the population at 40,000 and 50,000 generations.
## Warning: The following aesthetics were dropped during statistical transformation:
## colour, shape
## i This can happen when ggplot fails to infer the correct grouping structure in
## the data.
## i Did you forget to specify a `group` aesthetic or to convert a numerical
## variable into a factor?
## The following aesthetics were dropped during statistical transformation:
## colour, shape
## i This can happen when ggplot fails to infer the correct grouping structure in
## the data.
## i Did you forget to specify a `group` aesthetic or to convert a numerical
## variable into a factor?end = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives' & gen == 50000 & acron != 'ran')
end$Generation <- factor(end$gen)
mid = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives' & gen == 40000 & acron != 'ran')
mid$Generation <- factor(mid$gen)
mvc_p = ggplot(mid, aes(x = acron, y=pop_uni_obj, group = acron, shape = Generation)) +
geom_point(col = mvc_col[1] , position = position_jitternudge(jitter.width = .03, nudge.x = -0.05), size = 2, alpha = 1.0) +
geom_boxplot(position = position_nudge(x = -.15, y = 0), lwd = 0.7, col = mvc_col[1], fill = mvc_col[1], width = .1, outlier.shape = NA, alpha = 0.0) +
geom_point(data = end, aes(x = acron, y=pop_uni_obj), col = mvc_col[2], position = position_jitternudge(jitter.width = .03, nudge.x = 0.05), size = 2, alpha = 1.0) +
geom_boxplot(data = end, aes(x = acron, y=pop_uni_obj), position = position_nudge(x = .15, y = 0), lwd = 0.7, col = mvc_col[2], fill = mvc_col[2], width = .1, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 5)
) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=c(0,1))+
scale_colour_manual(values = c(mvc_col[1],mvc_col[2])) +
p_theme
plot_grid(
mvc_p +
ggtitle("Satisfactory trait coverage comparisons") +
theme(legend.position="none"),
legend,
nrow=2,
rel_heights = c(1,.05),
label_size = TSIZE
)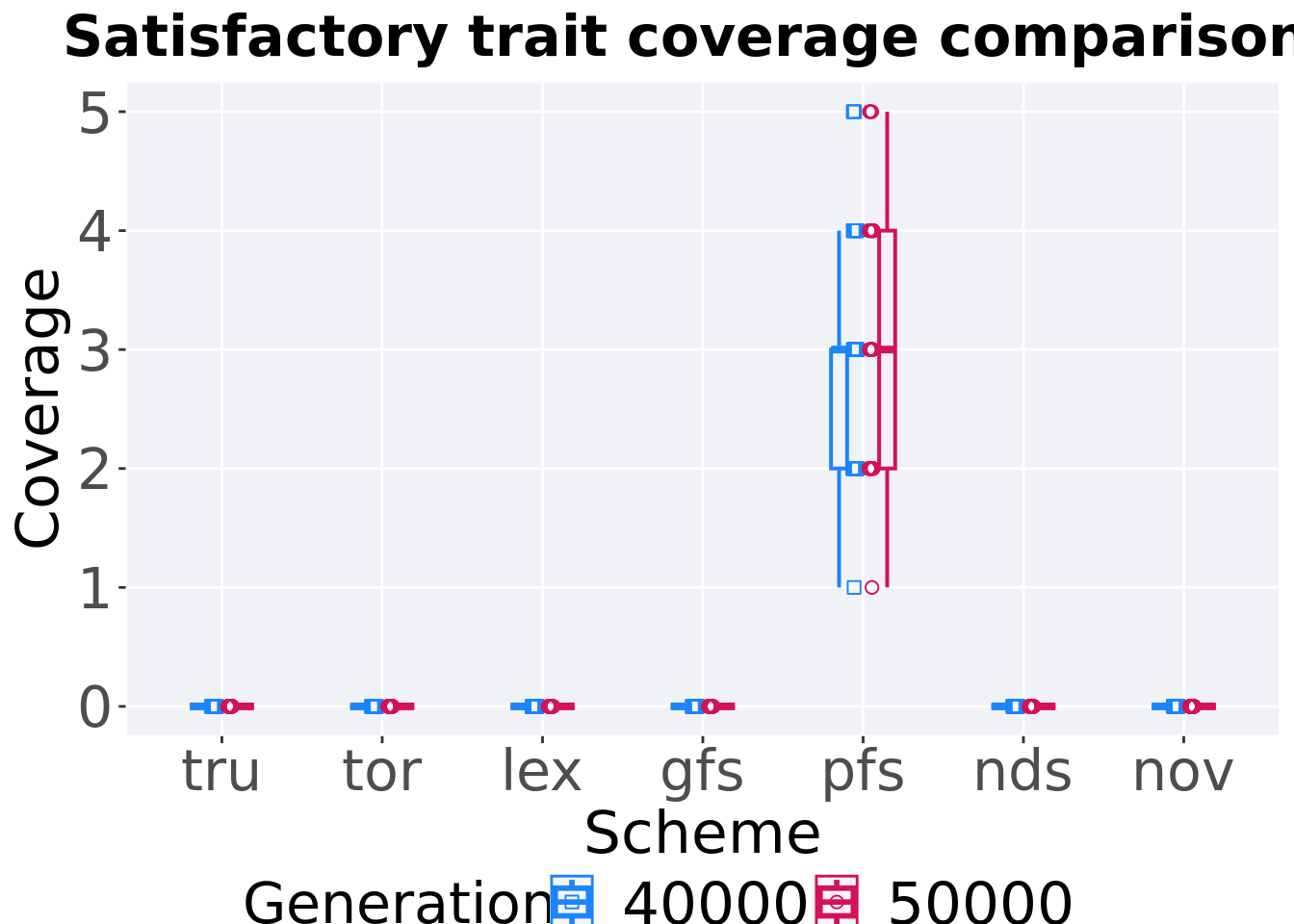
4.5.1.3.1 Stats
Summary statistics for the activation gene coverage at 40,000 and 50,000 generations.
slices = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives' & (gen == 50000 | gen == 40000) & acron != 'ran')
slices$Generation <- factor(slices$gen, levels = c(50000,40000))
slices$acron = factor(slices$acron, levels = c('pfs','nds', 'lex', 'gfs', 'tor', 'tru', 'nov', 'ran'))
slices %>%
group_by(acron, Generation) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(pop_uni_obj)),
min = min(pop_uni_obj, na.rm = TRUE),
median = median(pop_uni_obj, na.rm = TRUE),
mean = mean(pop_uni_obj, na.rm = TRUE),
max = max(pop_uni_obj, na.rm = TRUE),
IQR = IQR(pop_uni_obj, na.rm = TRUE)
)## `summarise()` has grouped output by 'acron'. You can override using the
## `.groups` argument.## # A tibble: 14 x 9
## # Groups: acron [7]
## acron Generation count na_cnt min median mean max IQR
## <fct> <fct> <int> <int> <int> <dbl> <dbl> <int> <dbl>
## 1 pfs 50000 50 0 1 3 3.14 5 2
## 2 pfs 40000 50 0 1 3 2.9 5 1
## 3 nds 50000 50 0 0 0 0 0 0
## 4 nds 40000 50 0 0 0 0 0 0
## 5 lex 50000 50 0 0 0 0 0 0
## 6 lex 40000 50 0 0 0 0 0 0
## 7 gfs 50000 50 0 0 0 0 0 0
## 8 gfs 40000 50 0 0 0 0 0 0
## 9 tor 50000 50 0 0 0 0 0 0
## 10 tor 40000 50 0 0 0 0 0 0
## 11 tru 50000 50 0 0 0 0 0 0
## 12 tru 40000 50 0 0 0 0 0 0
## 13 nov 50000 50 0 0 0 0 0 0
## 14 nov 40000 50 0 0 0 0 0 0Truncation selection comparisons.
wilcox.test(x = filter(slices, acron == 'tru' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'tru' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "tru" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "tru" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 0Tournament selection comparisons.
wilcox.test(x = filter(slices, acron == 'tor' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'tor' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "tor" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "tor" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 0Lexicase selection comparisons.
wilcox.test(x = filter(slices, acron == 'lex' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'lex' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "lex" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "lex" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 0Genotypic fitness sharing comparisons.
wilcox.test(x = filter(slices, acron == 'gfs' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'gfs' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "gfs" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "gfs" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 0Phenotypic fitness sharing comparisons.
wilcox.test(x = filter(slices, acron == 'pfs' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'pfs' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "pfs" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "pfs" & Generation == 40000)$pop_uni_obj
## W = 1423.5, p-value = 0.2118
## alternative hypothesis: true location shift is not equal to 0Nondominated sorting comparisons.
wilcox.test(x = filter(slices, acron == 'nds' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'nds' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "nds" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "nds" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 0Novelty search comparisons.
wilcox.test(x = filter(slices, acron == 'nov' & Generation == 50000)$pop_uni_obj,
y = filter(slices, acron == 'nov' & Generation == 40000)$pop_uni_obj,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "nov" & Generation == 50000)$pop_uni_obj and filter(slices, acron == "nov" & Generation == 40000)$pop_uni_obj
## W = 1250, p-value = NA
## alternative hypothesis: true location shift is not equal to 04.5.2 Activation gene coverage
Activation gene coverage analysis.
4.5.2.1 Coverage over time
Activation gene coverage over time.
lines = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives') %>%
group_by(`Selection\nScheme`, gen) %>%
dplyr::summarise(
min = min(uni_str_pos),
mean = mean(uni_str_pos),
max = max(uni_str_pos)
)## `summarise()` has grouped output by 'Selection Scheme'. You can override using
## the `.groups` argument.ggplot(lines, aes(x=gen, y=mean, group = `Selection\nScheme`, fill =`Selection\nScheme`, color = `Selection\nScheme`, shape = `Selection\nScheme`)) +
geom_ribbon(aes(ymin = min, ymax = max), alpha = 0.1) +
geom_line(size = 0.5) +
geom_point(data = filter(lines, gen %% 2000 == 0 & gen != 0), size = 1.5, stroke = 2.0, alpha = 1.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100")
) +
scale_x_continuous(
name="Generations",
limits=c(0, 50000),
breaks=c(0, 10000, 20000, 30000, 40000, 50000),
labels=c("0e+4", "1e+4", "2e+4", "3e+4", "4e+4", "5e+4")
) +
scale_shape_manual(values=SHAPE)+
scale_colour_manual(values = cb_palette) +
scale_fill_manual(values = cb_palette) +
ggtitle('Activation gene coverage over time')+
p_theme + theme(legend.title=element_blank(),legend.text=element_text(size=11)) +
guides(
shape=guide_legend(ncol=2, title.position = "bottom"),
color=guide_legend(ncol=2, title.position = "bottom"),
fill=guide_legend(ncol=2, title.position = "bottom")
)
4.5.2.2 Coverage comparison
Best activation gene coverage in the population at 40,000 and 50,000 generations.
mvc_p = ggplot(mid, aes(x = acron, y=uni_str_pos, group = acron, shape = Generation)) +
geom_point(col = mvc_col[1] , position = position_jitternudge(jitter.width = .03, nudge.x = -0.05), size = 2, alpha = 1.0) +
geom_boxplot(position = position_nudge(x = -.15, y = 0), lwd = 0.7, col = mvc_col[1], fill = mvc_col[1], width = .1, outlier.shape = NA, alpha = 0.0) +
geom_point(data = end, aes(x = acron, y=uni_str_pos), col = mvc_col[2], position = position_jitternudge(jitter.width = .03, nudge.x = 0.05), size = 2, alpha = 1.0) +
geom_boxplot(data = end, aes(x = acron, y=uni_str_pos), position = position_nudge(x = .15, y = 0), lwd = 0.7, col = mvc_col[2], fill = mvc_col[2], width = .1, outlier.shape = NA, alpha = 0.0) +
scale_y_continuous(
name="Coverage",
limits=c(0, 100),
breaks=seq(0,100, 20),
labels=c("0", "20", "40", "60", "80", "100") ) +
scale_x_discrete(
name="Scheme"
)+
scale_shape_manual(values=c(0,1))+
scale_colour_manual(values = c(mvc_col[1],mvc_col[2])) +
p_theme
plot_grid(
mvc_p +
ggtitle("Activation gene coverage comparisons") +
theme(legend.position="none"),
legend,
nrow=2,
rel_heights = c(1,.05),
label_size = TSIZE
)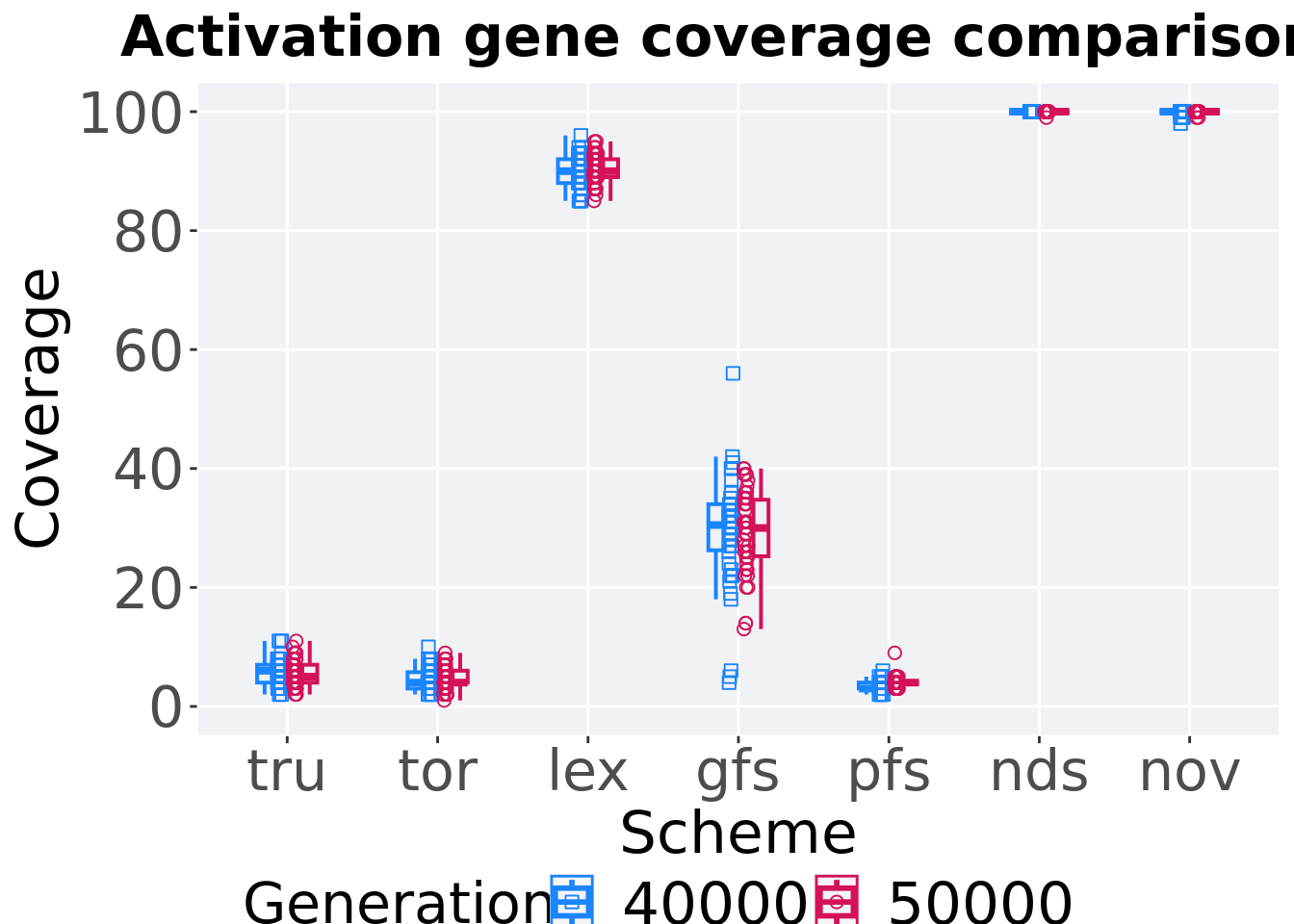
4.5.2.3 Stats
Summary statistics for the activation gene coverage at 40,000 and 50,000 generations.
slices = filter(cc_over_time_mvc, diagnostic == 'contradictory_objectives' & (gen == 50000 | gen == 40000) & acron != 'ran')
slices$Generation <- factor(slices$gen, levels = c(50000,40000))
slices$acron = factor(slices$acron, levels = c('nov','nds','lex','gfs','tor','tru','pfs'))
slices %>%
group_by(acron, Generation) %>%
dplyr::summarise(
count = n(),
na_cnt = sum(is.na(uni_str_pos)),
min = min(uni_str_pos, na.rm = TRUE),
median = median(uni_str_pos, na.rm = TRUE),
mean = mean(uni_str_pos, na.rm = TRUE),
max = max(uni_str_pos, na.rm = TRUE),
IQR = IQR(uni_str_pos, na.rm = TRUE)
)## `summarise()` has grouped output by 'acron'. You can override using the
## `.groups` argument.## # A tibble: 14 x 9
## # Groups: acron [7]
## acron Generation count na_cnt min median mean max IQR
## <fct> <fct> <int> <int> <int> <dbl> <dbl> <int> <dbl>
## 1 nov 50000 50 0 99 100 100. 100 0
## 2 nov 40000 50 0 98 100 99.8 100 0
## 3 nds 50000 50 0 99 100 100. 100 0
## 4 nds 40000 50 0 100 100 100 100 0
## 5 lex 50000 50 0 85 90 90.3 95 3
## 6 lex 40000 50 0 85 90 90.0 96 4
## 7 gfs 50000 50 0 13 30 29.4 40 9.5
## 8 gfs 40000 50 0 4 30.5 29.3 56 7.75
## 9 tor 50000 50 0 1 4 4.64 9 2
## 10 tor 40000 50 0 2 4 4.54 10 2.75
## 11 tru 50000 50 0 2 5 5.6 11 3
## 12 tru 40000 50 0 2 6 5.8 11 3
## 13 pfs 50000 50 0 3 4 4.02 9 0
## 14 pfs 40000 50 0 2 3 3.42 6 1Truncation selection comparisons.
wilcox.test(x = filter(slices, acron == 'tru' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'tru' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "tru" & Generation == 50000)$uni_str_pos and filter(slices, acron == "tru" & Generation == 40000)$uni_str_pos
## W = 1175, p-value = 0.6039
## alternative hypothesis: true location shift is not equal to 0Tournament selection comparisons.
wilcox.test(x = filter(slices, acron == 'tor' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'tor' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "tor" & Generation == 50000)$uni_str_pos and filter(slices, acron == "tor" & Generation == 40000)$uni_str_pos
## W = 1351.5, p-value = 0.4781
## alternative hypothesis: true location shift is not equal to 0Lexicase selection comparisons.
wilcox.test(x = filter(slices, acron == 'lex' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'lex' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "lex" & Generation == 50000)$uni_str_pos and filter(slices, acron == "lex" & Generation == 40000)$uni_str_pos
## W = 1321.5, p-value = 0.6221
## alternative hypothesis: true location shift is not equal to 0Genotypic fitness sharing comparisons.
wilcox.test(x = filter(slices, acron == 'gfs' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'gfs' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "gfs" & Generation == 50000)$uni_str_pos and filter(slices, acron == "gfs" & Generation == 40000)$uni_str_pos
## W = 1223.5, p-value = 0.8575
## alternative hypothesis: true location shift is not equal to 0Phenotypic fitness sharing comparisons.
wilcox.test(x = filter(slices, acron == 'pfs' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'pfs' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "pfs" & Generation == 50000)$uni_str_pos and filter(slices, acron == "pfs" & Generation == 40000)$uni_str_pos
## W = 1709, p-value = 0.0006733
## alternative hypothesis: true location shift is not equal to 0Nondominated sorting comparisons.
wilcox.test(x = filter(slices, acron == 'nds' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'nds' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "nds" & Generation == 50000)$uni_str_pos and filter(slices, acron == "nds" & Generation == 40000)$uni_str_pos
## W = 1225, p-value = 0.3271
## alternative hypothesis: true location shift is not equal to 0Novelty search comparisons.
wilcox.test(x = filter(slices, acron == 'nov' & Generation == 50000)$uni_str_pos,
y = filter(slices, acron == 'nov' & Generation == 40000)$uni_str_pos,
alternative = 't')##
## Wilcoxon rank sum test with continuity correction
##
## data: filter(slices, acron == "nov" & Generation == 50000)$uni_str_pos and filter(slices, acron == "nov" & Generation == 40000)$uni_str_pos
## W = 1476, p-value = 0.007657
## alternative hypothesis: true location shift is not equal to 0